Поскольку терапевтические возможности против коронавируса были сосредоточены главным образом на блокировании взаимодействия между его спайковым белком и рецептором ACE2 на клетках хозяина, SARS-CoV-2 имеет несколько дополнительных критических белков, которые потенциально могут быть нацелены на препараты, уже одобренные для использования против других вирусов. Одним из таких вирусных белков является основная протеаза (Mpro), которая необходима для разделения новоиспеченных полипептидов на их функциональные составные части.
Ученые из отдела биофизики Всеиндийского института медицинских наук недавно провели несколько многообещающих ингибиторов Mpro через шаги, чтобы увидеть, что прилипает. Их результаты, опубликованные в журнале Американского химического общества ACS Omega, свидетельствуют о том, что лекарство от ВИЧ, известное как кобицистат, выглядит довольно хорошо. Но что именно представляет собой" хороший " препарат здесь, и как исследователи вообще находят его?
Хорошая отправная точка - иметь некоторое представление о том, как выглядит ваша цель. В этом случае 3-D структура Mpro недавно стала доступной, чтобы служить основой для того, что известно в бизнесе как рациональный структурный дизайн лекарств. Чтобы начать работу, исследователи практически проверили библиотеку одобренных лекарственных соединений в базе фармацевтических знаний Банка лекарств, чтобы найти возможные ингибиторы Mpro. Как правило, это включает в себя проведение молекулярных докинговых исследований, чтобы составить короткий список лучших кандидатов. Одна из наиболее распространенных метрик, используемых в этой работе, заключается в поиске молекул с высоким счетом стыковки скольжения и энергией скольжения.
GlideScore вычисляется с помощью программного обеспечения, такого как Glide от Schrodinger. Короче говоря, он ранжирует так называемые "позы" различных лигандов, имитируя свободную энергию связывания; чем более отрицательное значение, тем плотнее связывание. Эта эмпирическая функция оценки включает термины для вкладов силового поля (электростатические, ван-дер-Ваальсовые), а также термины, вознаграждающие или наказывающие другие взаимодействия, которые, как известно, влияют на связывание лиганда. Как правило, можно смоделировать до 300 атомов и 50 вращающихся связей, что достаточно для низкомолекулярных лекарств или даже пептидных лигандов примерно до 11 остатков.
Авторы также использовали более продвинутый метод расчета энергии связи лиганд-белковых комплексов, известный как молекулярная механика-площадь поверхности Пуассона-Больцмана (MM-PBSA). Этот метод сочетает энергетические расчеты, основанные на молекулярной механике, с расчетами свободной энергии, основанными на неявных моделях растворителей. Другими словами, он оценивает свободную энергию связывания лиганд-белкового комплекса как разность между свободной энергией комплекса и свободными энергиями несвязанных компонентов, включая как энтропийные, так и энтальпийные члены.
Следующий шаг в процессе открытия лекарств обычно заключается в более детальном молекулярно-динамическом моделировании, чтобы дополнительно распределить молекулы-кандидаты и уменьшить количество, которое необходимо экспериментально проверить. Моделирование MD уточняет взаимодействие состыкованных комплексов, обеспечивая гибкость белка и детальные эффекты растворителя. Результаты моделирования МД представлены в виде графиков RMSD (среднеквадратичного отклонения) флуктуаций атомного положения белкового остова, или радиуса вращения, а также числа водородных связей, как функции длины пробега, заданной в наносекундах.
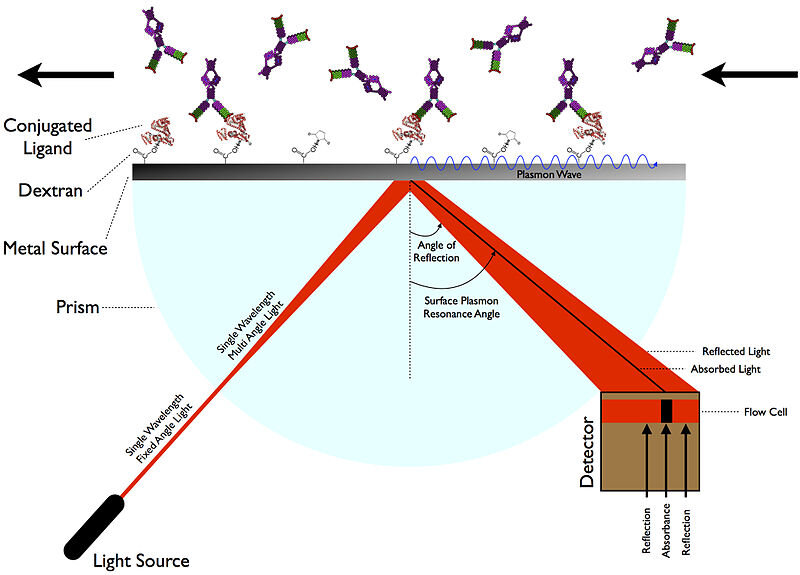
Когда все сказано и сделано, то, что авторы действительно ищут,-это фактическая истинная Кинетика молекулярного взаимодействия выбранных ими молекул. Только реальные измерения могут обеспечить это, и, как мы обсуждали несколько дней назад, есть несколько новых видов инструментов, которые могут это сделать. Возможно, наиболее удобным методом определения кинетики молекулярного взаимодействия является SPR (поверхностный плазмонный резонанс). SPR доставляет сенсорграммы, из которых можно определить константы скорости ассоциации (kon) и константы скорости диссоциации (koff) для связывания потенциального лекарственного ингибитора с мишенью, такой как Mpro. У одного производителя инструментов SPR, Nicoya, есть отличный пост в блоге, который более подробно описывает, как работают эти анализы.
Авторам удалось определить равновесные константы диссоциации, KD (M), для потенциальных ингибиторов типа кобицистата, так как из кинетических данных легко вывести соотношение: KD = koff/kon. Единственное, что оставалось сделать после идентификации кобицистата как победителя первичного кандидата на препарат, - это проверить, что он ингибирует Mpro в анализе активности фермента. Для препаратов, которые потенциально могут блокировать взаимодействие между спайковым белком SARS-CoV-2 и его рецептором ACE2, ингибитор может быть применен к анализу SPR в качестве "разрушителя" связывания лиганда с мишенью. Кинетические данные в такой ситуации получить было бы нелегко.
Поскольку мишень Mpro не была перенесена в SPR-дружественное приложение, авторы измерили ингибирование активности Mpro с помощью универсального протеазного анализа. Они смогли получить значение IC50 для кобицистата ≈6,7 мкм, что было более благоприятным, чем для других потенциальных препаратов кангрелора и денуфосола (0,9 мм и 1,3 мм соответственно). IC50 показывает, сколько препарата необходимо для ингибирования активности на 50%.
Было также обнаружено, что участок расщепления, в котором действует Mpro, отличается от участка расщепления, в котором действуют многие существующие протеазы человека. Это к счастью, потому что любой препарат, который блокирует наши собственные протеазы, несомненно, будет иметь значительные побочные эффекты. Другая протеаза SARS-CoV-2, PLpro, распознает важную молекулу, известную как убиквитин, и любые попытки ингибировать эту протеазу могут привести к разрушению наших собственных критических деубиквитиназных систем.
В духе победы над всеми вещами COVID произошли некоторые дальнейшие интересные события в более широкой сфере готовности вакцин, а именно в расшифровке того, как и, следовательно, можно ли ожидать, что эти новые вакцины будут работать. Берт Хьюберт возглавил общественное обвинение, чтобы попытаться выяснить, как Pfizer оптимизировала свою мРНК-вакцину для трансляции в наших клетках. В частности, он создал задачу программирования, чтобы найти предполагаемый алгоритм, с помощью которого была выполнена кодонная оптимизация вакцины длиной ~4000 символов.
Это предполагает, конечно, что был сделан какой-то реальный алгоритм, а не просто ручная обработка отдельных кодонов, и в этом случае самый короткий алгоритм вообще не был бы очень коротким. На самом деле это был бы почти тот же самый алгоритм, который создал всю Вселенную, которая эволюционировала в Pfizer. Последние обновления, которые включают в себя представленные пользователями алгоритмы, предсказывающие фактические кодоны с точностью более 90% от зарождающегося сообщества хакеров, ставших биологами, можно найти на сайте обратного инжиниринга вакцины Pfizer Берта.
В заключение следует отметить, что другие конкурирующие мРНК-вакцины, такие как Moderna или CureVac, скорее всего, будут очень похожи на вакцину Pfiver. Например, парень по имени Павол Руснак только что извлек последовательность CureVac в текстовый файл и разместил ее здесь. Хотя эти кодоны кодируют почти те же аминокислоты, что и BNT162b2, 33% кодонов отличаются. Хотя может быть трудно отслеживать все, что происходит с более крупной экосистемой SARS-CoV-2, один невероятный технический источник не должен ускользнуть от внимания ни одного серьезного наблюдателя, и это аккаунт в социальных сетях Ersa Flavinkins, он же @flavinkins. Обязательно настраивайтесь на все последние факты, теории, домыслы и, конечно же, заговоры. | |
| Просмотров: 511 | |